The Annie Huang Lab has broad interests in the molecular pathogenesis of paediatric brain tumours with an aim of improving diagnosis and treatment. Our studies are particularly focused on rare malignant brain tumours. These are “orphan diseases”, primarily arising in infants or younger children, a population most vulnerable to toxicities of conventional multi-modal oncologic therapy.
We work to understand biological determinants and mechanisms of disease phenotypes, including tumour invasion and metastases – key factors driving treatment associated morbidity and mortality. Our end goal is to translate basic molecular knowledge to refine understanding of specific disease phenotypes and devise innovative, less toxic biology-driven approaches to cure infants and young children with malignant brain tumours. Our disease area of interests include ATRTs, ETMRs and Pineoblastoma which make up the majority of malignant infant embryonal brain tumours.
Over the last 10-15 years we have exploited the unique biomaterials and clinical data from the Rare Brain Tumor Consortium registry and biorepository to map the unknown molecular landscapes of these very rare diseases. Together with our large network of collaborators, we are now exploiting this critical groundwork to identify drivers of malignant and clinical phenotypes within and across the different infant rare brain tumours by applying functional epigenomics, genome-wide functional screens and single cell methodologies.
Our research is split into three broad categories:
 ATRTs are malignant brain tumours resulting from bi-allelic loss of function mutations in the SMARCB1 gene, which encodes BAF47, a critical component of the SWI/SNF remodeling complex. Although ATRTs have simple genomes with few other mutations, clinical presentation and response to treatment can be quite variable and unpredictable. Recent studies by our lab and others show ATRTs comprise at least 3 molecular sub-groups. Current projects in the lab aims to understand the role of non-coding genes in ATRTs and how they contribute to ATRT phenotypes. In addition, we are combining single cell analyses of human ATRTs, hNSC derived as well as tumour cell models to determine the role of the tumour micro-environment in ATRT development. We also undertake studies in collaboration with the laboratory of Dr James Rutka to identify clinically useful therapeutic compounds and create animal models for ATRTs.
ATRTs are malignant brain tumours resulting from bi-allelic loss of function mutations in the SMARCB1 gene, which encodes BAF47, a critical component of the SWI/SNF remodeling complex. Although ATRTs have simple genomes with few other mutations, clinical presentation and response to treatment can be quite variable and unpredictable. Recent studies by our lab and others show ATRTs comprise at least 3 molecular sub-groups. Current projects in the lab aims to understand the role of non-coding genes in ATRTs and how they contribute to ATRT phenotypes. In addition, we are combining single cell analyses of human ATRTs, hNSC derived as well as tumour cell models to determine the role of the tumour micro-environment in ATRT development. We also undertake studies in collaboration with the laboratory of Dr James Rutka to identify clinically useful therapeutic compounds and create animal models for ATRTs.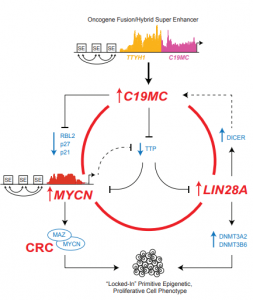 ETMR is a newly recognized rare brain tumour previously considered many different diagnoses until the discovery of C19MC genomic amplification/fusions as a specific marker for this group of diseases. Our studies have shown that this large primate specific and ES cell enriched miRNA cluster, is a major oncogenic driver of the highly malignant and aggressive nature of ETMRs. Current projects in the lab aims to define the specific contribution of C19MC and other related oncogenes LIN28A and MYCN to ETMR phenotypes. We aim to exploit this knowledge to develop therapeutic models, and identify targetable vulnerabilities in ETMR therapeutics.
ETMR is a newly recognized rare brain tumour previously considered many different diagnoses until the discovery of C19MC genomic amplification/fusions as a specific marker for this group of diseases. Our studies have shown that this large primate specific and ES cell enriched miRNA cluster, is a major oncogenic driver of the highly malignant and aggressive nature of ETMRs. Current projects in the lab aims to define the specific contribution of C19MC and other related oncogenes LIN28A and MYCN to ETMR phenotypes. We aim to exploit this knowledge to develop therapeutic models, and identify targetable vulnerabilities in ETMR therapeutics. he MYC/N family of oncogenes are potent drivers of tumourigenesis which are often de-regulated in aggressive sub-groups of different paediatric embryonal brain tumours including ATRT, ETMR, pineoblastoma and medulloblastoma. We are interested in understanding the cellular consequences of MYC/N deregulation in embryonal tumours. Ongoing projects in the lab include defining MYC/N driven pathways in different tumour models, and examining the specific mechanisms by which MYC/N specifies invasive and metastatic phenotypes in the different classes of rare and infant brain tumours. The end goal of these studies is to better predict and treat MYC/N driven embryonal brain tumours.
he MYC/N family of oncogenes are potent drivers of tumourigenesis which are often de-regulated in aggressive sub-groups of different paediatric embryonal brain tumours including ATRT, ETMR, pineoblastoma and medulloblastoma. We are interested in understanding the cellular consequences of MYC/N deregulation in embryonal tumours. Ongoing projects in the lab include defining MYC/N driven pathways in different tumour models, and examining the specific mechanisms by which MYC/N specifies invasive and metastatic phenotypes in the different classes of rare and infant brain tumours. The end goal of these studies is to better predict and treat MYC/N driven embryonal brain tumours.