Our research involves a wide range of reagents, methods, experimental approaches and technologies that are continually changing and advancing. Researchers at the SickKids RI are fortunate in having access to specialized facilities that provide advanced analytical capabilities, such as the SickKids proteomics, analytics, robotics and chemical biology centre (SPARC), and the Imaging Facility with state-of-the art optical and fluorescence microscopes. We also have access to a wide range of cutting-edge expertise and other support critical to research success. What follows are a few examples where access to world-class technical support has been critical to our research progress.
Genetic analysis used to take weeks or months and cost thousands of dollars, now it takes days or hours and costs much less. We have exploited ongoing advances in this field in several studies, including one that defined the genetic cause of a known hereditary condition: gray platelet syndrome (GPS), and one that discovered a new disease: ARPC1B deficiency.
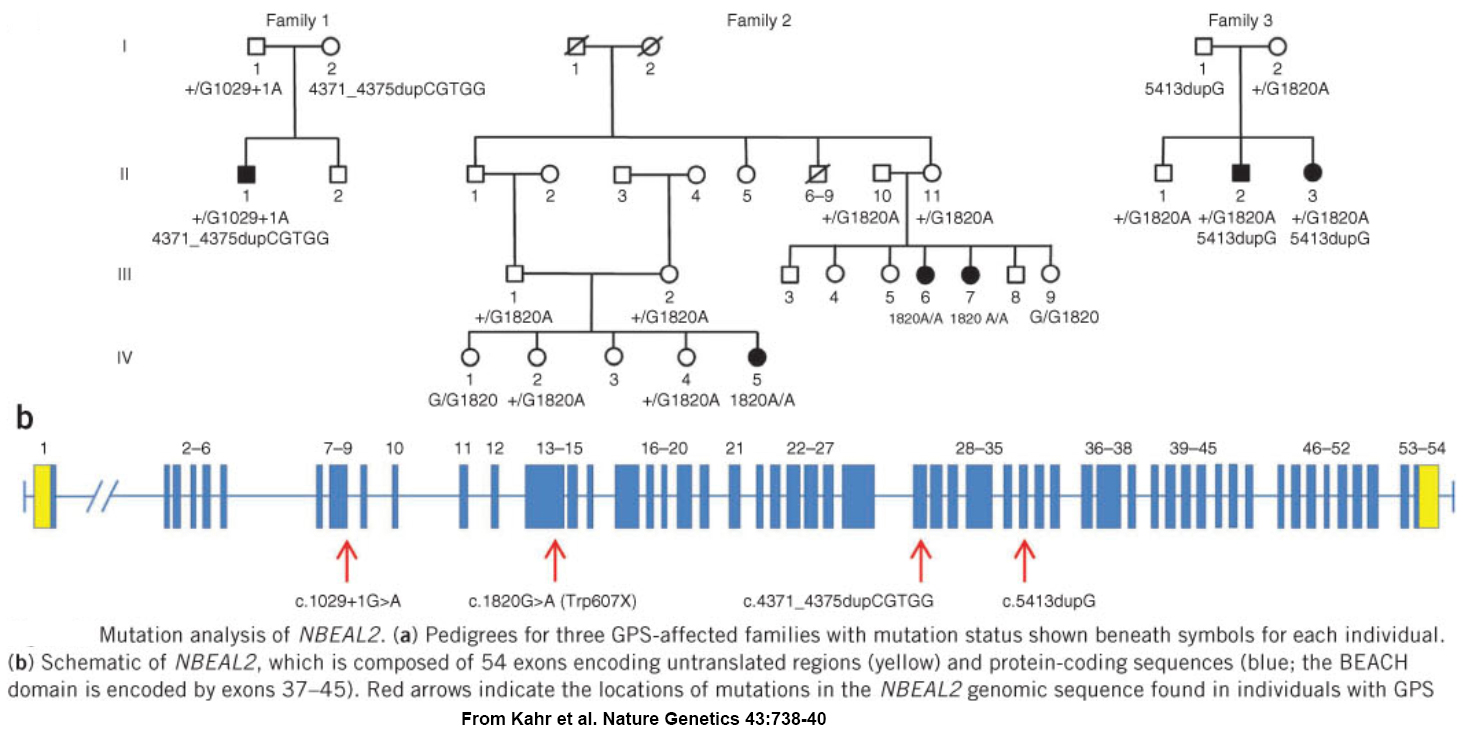
GPS was first reported in 1971, and a worldwide search for a cause was well underway when our relatively small international collaborative group became involved. We used homozygosity mapping of a chip-based genome-wide human single nucleotide polymorphism (SNP) array to localize the GPS locus to a 1.7 megabase region of chromosome 3 containing over 70 genes. Sequencing all of them to find mutations would have taken a considerable effort, so instead we employed “next-generation sequencing” to analyzed mRNA from platelets – which may not have nuclei but they are produced by megakaryocytes which abundantly express RNA. We detected a transcript that was missing in GPS patients, and as a result we were one of 3 groups to simultaneously report loss of NBEAL2 expression as the molecular cause of GPS.
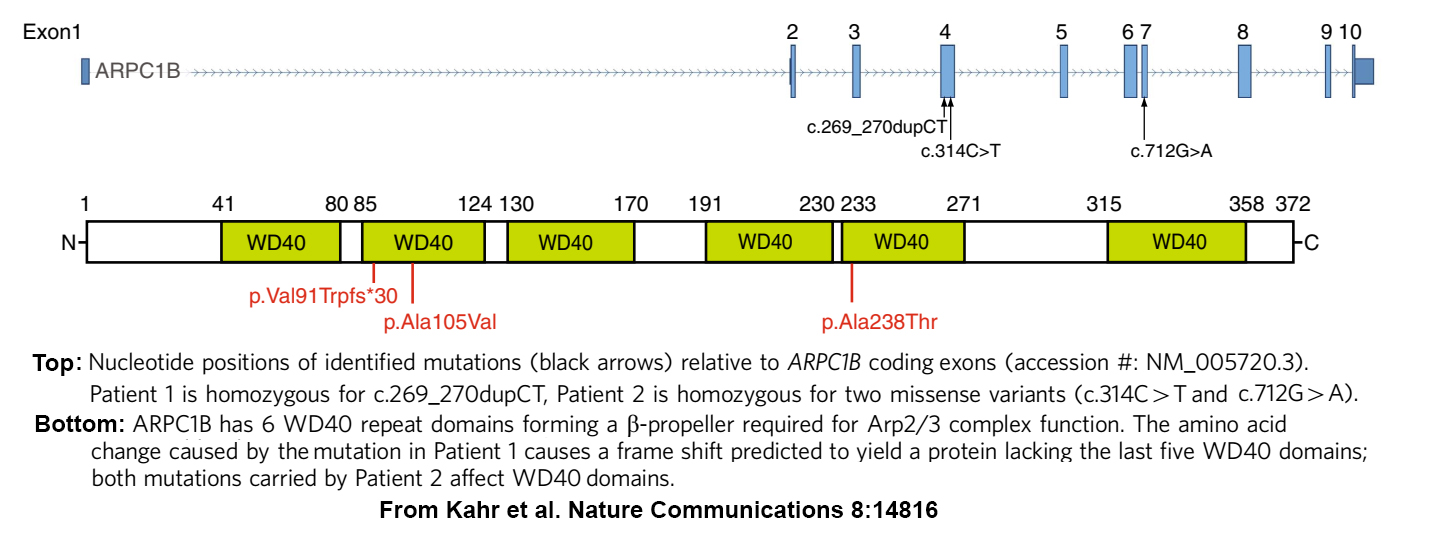
A few years later, we joined several SickKids colleagues in an effort to determine what was wrong with an especially challenging patient (see home page). At the time genomics analysis had reached the point where many hereditary disorders, including those affecting immunity/inflammation, platelets and the gastrointestinal system, had been linked to panels of genes where pathogenic and predisposing variants had been identified, or were predicted to occur. Unfortunately, scans of those panels for variants in our index patient consistently turned up negative. So eventually we used a less targeted approach and undertook a Whole Exome Sequence (WES) analysis of every gene expressed by the patient and his parents (and by another suspected patient and his parents). This search detected homozygosity for potentially pathogenic ARPC1B variants in both patients, for whom reduced or absent ARPC1B expression was confirmed by platelet protein analysis.
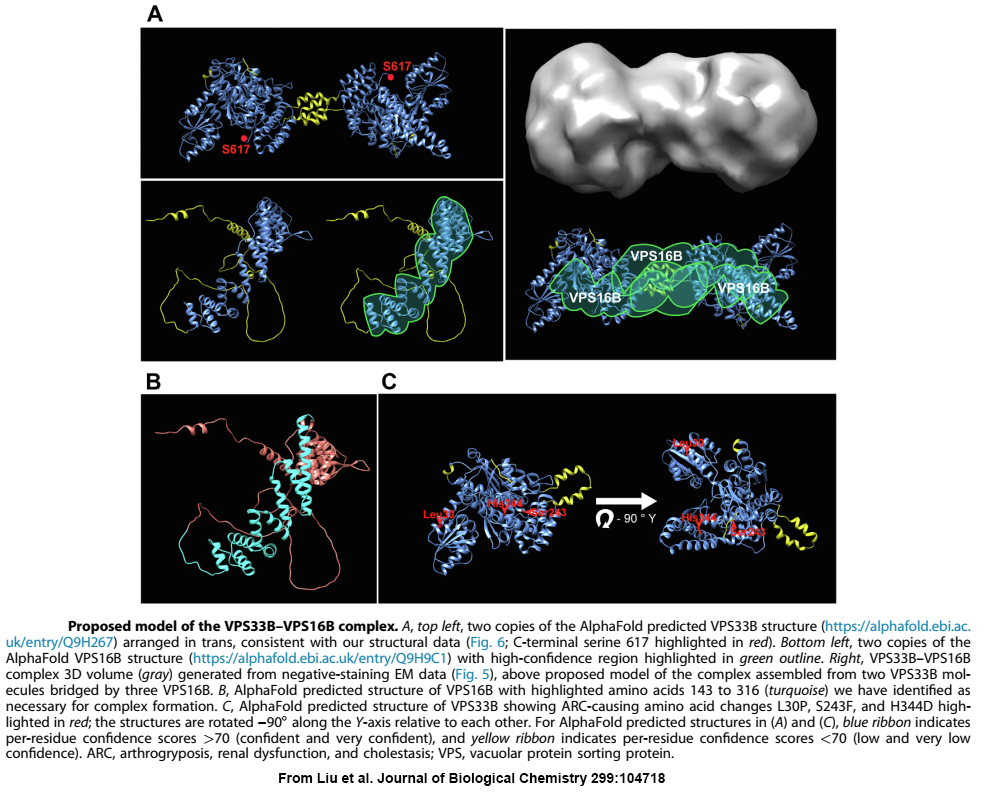
When we first linked VPS33B and VPS16B to platelet a-granule production and found these proteins formed a complex of unknown function, the obvious next step was to determine the structure of VPS16B-33B. At the time, the established way of doing this was to prepare milligram quantities of purified protein/complex, use it to form crystals, and perform X-ray diffraction analysis. Working with VPS16B-33B turned out to be challenging: we couldn’t make much of it, and it was not keen on forming crystals. Fortunately, other analytical options were available, including high-resolution cryo-electron microscopy (cryo-EM) established at the SickKids Nanoscale Biomedical Imaging Facility by our collaborator John Rubinstein. Using microgram amounts of protein we were able to use cryo-EM and other analytical techniques to determine the overall shape and size of the VPS16B-33B complex, and determine the number of components and their orientations. We also made use of recent advances in artificial intelligence by employing AlphaFold to obtain predicted protein structures that we were able to fit into the envelope of VPS16B-33B.