Dr. McGlade’s research program is directed towards understanding the specificity and dynamics of protein-protein interactions involved in cell signalling events that contribute to the development of cancer. Work in her laboratory focuses on the molecular structure, cellular functions and interaction networks of modular adaptor proteins that regulate critical oncogenic signalling pathways, tumour progression and metastasis. Together with clinical and industry collaborators they work towards translating knowledge of specific molecular signalling events into strategies for drug discovery. Ongoing projects in the laboratory focus on how cell type specific expression of modular adaptors and isoforms promotes the activation of unique oncogenic signalling pathways, regulation of ubiquitin ligase activity, and investigation of the oncogenic signalling pathways that control alternative splicing in tumour cells. The long-term goal of her research is to advance knowledge of the molecular mechanisms that regulate signal transduction and determine the specific molecular events that can be developed into targets for therapeutic intervention.
Research
Current projects

Alternative splicing of pre-mRNA is a key mechanism for generating proteome diversity by creation of mature mRNAs that encode distinct protein products. Misregulation of alternative splicing and the associated changes in protein isoform expression is an important mechanism that underlies cancer initiation, progression and drug resistance. Splicing factor mutations, misexpression, or post-translational modifications in cancer cells lead to changes in spliceosome activity and dysregulated splicing of genes in critical signaling pathways, including promoting expression of tumourigenic protein isoforms. Targetting splicing regulators through antisense oligonucleotides and small molecule inhibitors of aberrantly activated splicing factors have been developed as therapeutics for cancer and other human diseases.
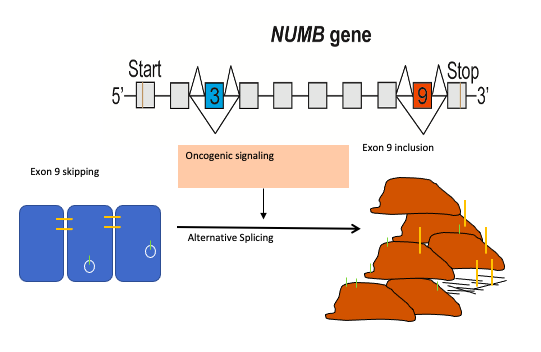
Numb is an endocytic adaptor protein that negatively regulates membrane receptors and adhesion molecules by regulating their endocytosis and lysosomal sorting. The central role of Numb in the regulation of multiple key signaling pathways led to the hypothesis that loss of Numb activity contributes to cancer development. Over the last decade evidence has accumulated that a switch in Numb alternative splicing, giving rise to the production of distinct protein isoforms, is a frequent occurrence in cancer and that increased inclusion of Numb exon 9 is a feature of many cancer types including lung, breast and colon cancer. Thus we proposed that targeting the mechanisms that regulate Numb splicing can be utilized to reverse the pathogenic activity of Numb exon9 expression.
The mechanisms by which dysregulation of the splicing machinery that regulates exon 9 is unknown. To address this, we designed and constructed dual fluorescent and dual luminescent mini-gene reporters that are able to mimic the splicing of Numb, and conducted kinase inhibitor and genome-wide KO screens to identify the pathways and factors involved. Our results are expected to define the developmental and oncogenic signaling networks that control the alternative splicing of NUMB as well as a program of coordinately regulated splicing events in cancer cells. Ongoing work will utilize this approach in high throughput screens to identify unique small molecules that specifically inhibit cancer associated splicing events.
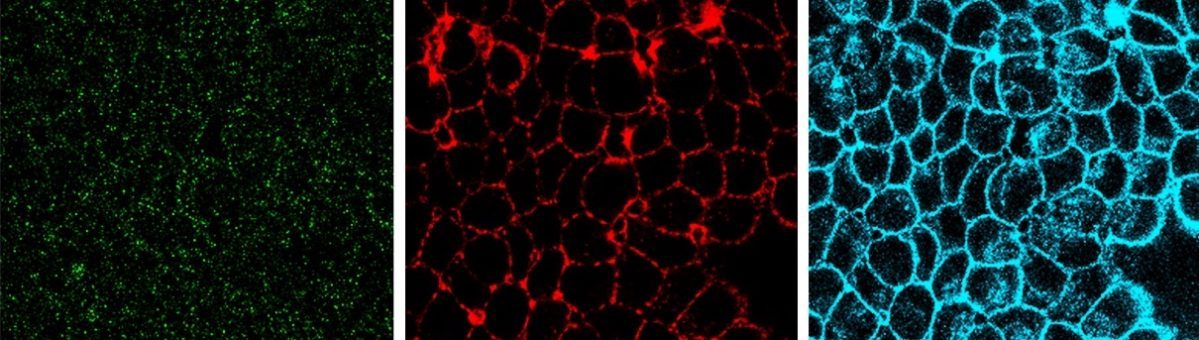
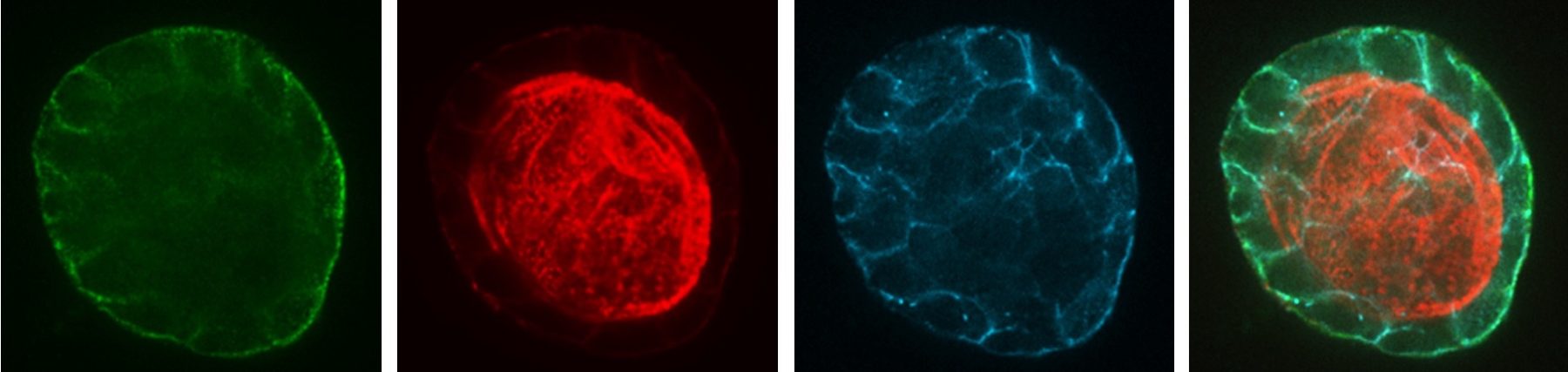
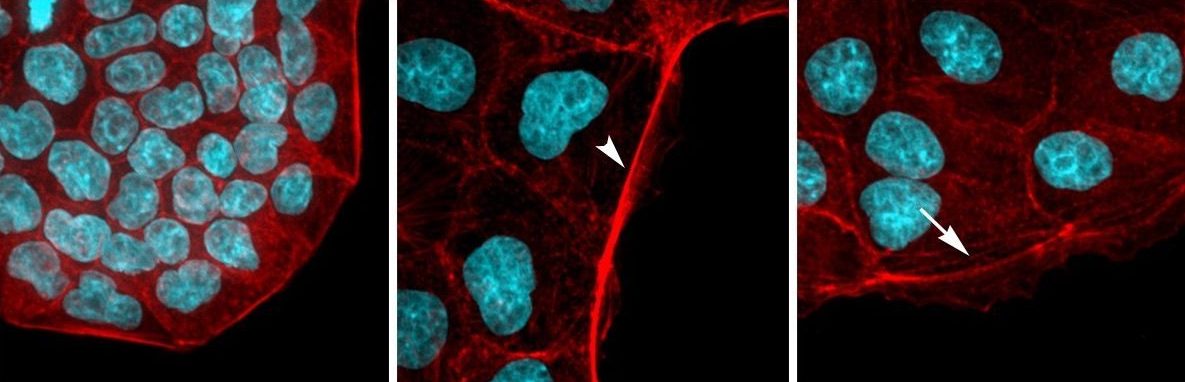
Endocytosis is the process of internalizing cell surface molecules, membrane lipids and extracellular material that serves to maintain cellular homeostasis, establish and maintain cell polarity and regulate signal transduction. Endocytosis is also a critical mechanism to bring about the dynamic changes in cell morphology and physiology associated with EMT and cancer progression. Numb is an endocytic adaptor protein with an amino terminal Phosphotyrosine Binding (PTB) domain shown to mediate interactions with the cytoplasmic domains of membrane receptors including Notch, integrins and E-cadherin, as well as a carboxy-termainal region with DPF tripeptide motifs that bind the -adaptin subunit of AP2 mediating localization to clathrin coated pits and an EH (Eps15 homology) domain-binding motif (NPF) that binds Eps15 and the EHD family endocytic proteins. Numb regulation of many pathways is linked to its role in endocytosis and trafficking.
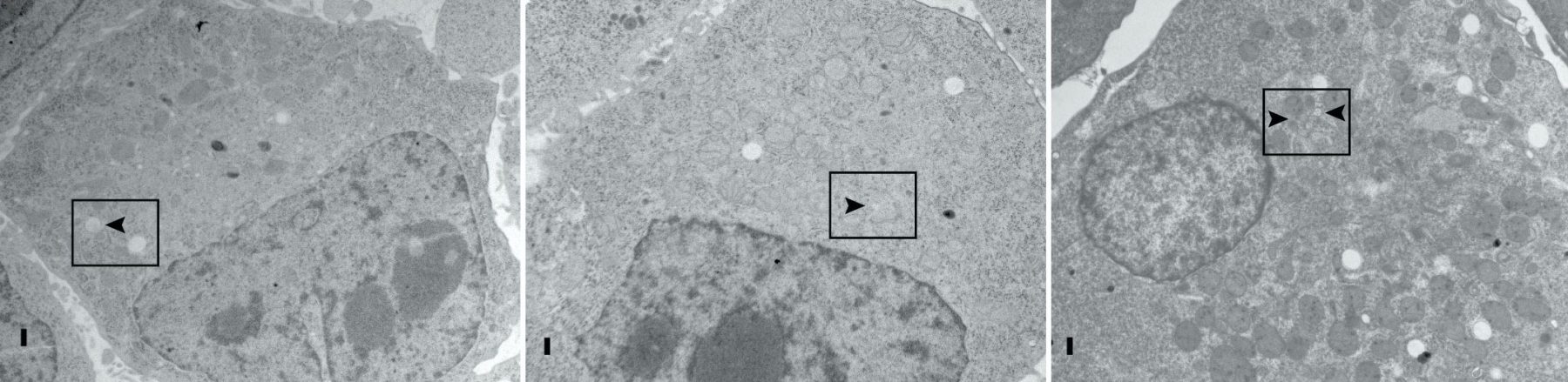
We proposed that alternative splicing of exon 9 in cancer acts as a switch that disrupts the normal function of Numb in endocytosis and vesicle trafficking to promote tumour progression and metastasis. To study differential Numb isoform function, the genomic region containing exon 9 was deleted from breast cancer cell lines and we utilized an unbiased quantitative proteomics approach to compare the total proteomes of control and targeted Ex9-deleted cell pools by quantitative mass spectrometry using Tandem Mass Tag (TMT) labeled peptides. We showed that the exon 9 containing protein isoform (exon9in) promotes breast cancer metastasis, causes remodeling of the endocytic network as well as increased abundance of of a set of growth factor receptors and adhesion molecules. We propose that changes in the balance of Numb isoform expression disrupts the post-endocytic down regulation of cancer associated growth factor receptors and adhesion molecules to drive EMT and tumour progression. On going studies will determine the effects of exon 9 inclusion on the internalization, recycling, degradation and signaling activity of cancer associated membrane receptors that are regulated by Numb isoform expression. In addition, we are investigating how the exon 9 encoded sequence alters interactions with the endocytic machinery as well as endocytic compartment localization.
Approaches to regulate CBL ubiquitin ligase activity and control oncogenic tyrosine kinase signaling
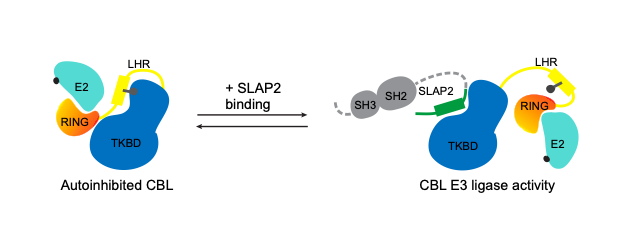
c-CBL (CBL) is an E3 ubiquitin ligase that attenuates signaling from many tyrosine kinase (TK) pathways by facilitating the ubiquitination of these proteins. Dysregulation of TK signaling is a major driver of oncogenesis. CBL mutations have been observed in a number of myeloid leukemias where they disrupt CBL regulation and lead to its inactivation. The loss of CBL E3 activity allows aberrant TK signaling to persist, contributing to uncontrolled cell proliferation in these cancers. CBL E3 activity is typically regulated through tyrosine phosphorylation, causing CBL to change from a closed autoinhibited form, to an open and active conformation. Subsets of oncogenic CBL mutations cause CBL to favour its closed conformation
The Src-Like Adaptor Proteins, SLAP and SLAP2, are required for CBL-dependent downregulation of multiple TK pathways in hematopoietic cells. SLAP and SLAP2 are thought to play a role in CBL recruitment to activated receptor complexes and our recent biochemical analyses have examined the effects of SLAP proteins on CBL E3 ligase activity and mapped the interaction interface. We proposed that SLAP dependent CBL ubiquitin ligase regulation can be exploited to modulate CBL in the context of oncogenic TK signaling. Our structural and in vitro analysis of the interaction between SLAP2 and CBL suggested a novel strategy to activate CBL through a protein-protein interaction induced conformational change. Based on this information we developed a high-throughput in vitro assay to screen for CBL activators. In order to characterize the ubiquitin signaling network effects of the SLAP-CBL interaction, we are also using proximity dependent biotinylation coupled with mass spectrometry. Future studies will investigate whether modulation of CBL ubiquitin ligase function has potential for therapeutic benefit cancers with elevated TK activity.
The use of tyrosine kinase inhibitors as front-line therapy of Ph+ acute lymphoblastic leukemia and chronic myeloid leukemia has led to improved outcome. However, drug resistance has become a major clinical issue and there is thus a critical need to better understand downstream signaling pathways that drive BCR-ABL positive leukemia and identify alternative drug targets.
The oncogenic potential of BCR-ABL relies on the constitutive activation of the ABL tyrosine kinase that phosphorylates tyrosine residues on protein substrates. Protein networks and signaling cascades made up of very specific and regulated protein-protein interactions are then activated, at the origin of the phenotypic properties of leukemic cells. The autophosphorylation of tyrosine 177 (Y177) in BCR-ABL has been shown to be essential. It leads to the SH2 domain-mediated recruitment of the GRB2 adaptor protein which through its SH3 domains binds to specific proteins and brings them into a complex with BCR-ABL. We identified a distinct adaptor protein called GADS (GRB2-related Adaptor Downstream of Shc) that is hematopoietic specific and also binds BCR-ABL on Y177. In prior work we utilized GADS knock out mice in bone marrow transduction/transplantation experiments to demonstrate a role for GADS in the latency and phenotype of BCR-ABL driven myeloproliferative disease. Since the SH3 domains of GADS have distinct binding specificity, we hypothesized that GADS expression in BCR-ABL driven leukemia activates distinct signaling pathways that impact leukemic cell survival, niche interactions and disease progression.

To study GADS specific signaling events in Ph+ leukemia we generated human GADS KO leukemia cell lines using CRISPR gene editing and compared the phosphoproteome in parental and GADS KO cells using a multiplexed quantitative phosphoproteomic approach. In addition, with the identification of endogenous GADS associated protein complexes in leukemia cells, we defined a novel BCR-ABL signaling axis. Ongoing studies are focused on the the functional characterization of GADS signaling and analysis in primary leukemia patient samples.
This project has been supported by grants from CIHR, Cancer Research Society and The Leukemia & Lymphoma Society of Canada (LLSC).
Special thanks to our funding!


