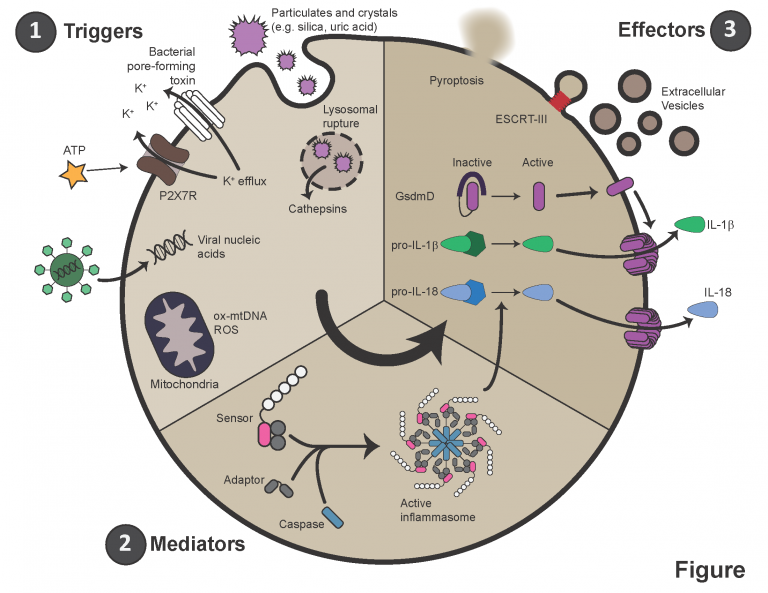
Pulmonary arterial hypertension (PAH) is characterized by immune infiltration of the peri-vascular space, elevated levels of circulating cytokines, and autoimmunity. Specifically, circulating levels of IL-1β and IL-6 are elevated in most patients and animal models of PAH. Secretion of IL-1β largely occurs downstream of a multi-protein complex termed the inflammasome. Activated by a variety of intra- and extra-cellular stresses or pathogens, inflammasomes aggregate and recruit caspases, which are serine proteases. These caspases then cleave pro-IL-1β into its mature form. They also cleave a protein called gasdermin D (GSDMD), which then oligomerizes and forms a large pore in the plasma membrane. IL-1β leaves the cell through this pore, and sometimes, this pore results in the death of the host cell, termed pyroptosis.
Our lab and others have shown that inflammasomes are activated in PAH. We are actively studying the mechanism of this activation, the cells in which it occurs, and when it arises in the context of disease. This work is providing critical information for further study and, importantly, inflammasome pathways are novel druggable targets in PAH.
Patients with connective tissue diseases (Scleroderma, Lupus, and others) are at a very high risk of developing PAH. Additionally, their prognosis and response to treatment is much poorer than those with idiopathic disease. This area is an intensive focus of research in order to improve outcomes for this high risk cohort. Robust animal models are lacking for the study of PAH in CTD patients. We have recently described a mouse model of CTD-PAH, and are currently using it to understand the interface between the extracellular matrix, the immune system, and the lung vasculature, to better understand this important disease.