Rare Neurodegenerative Disease: At the Molecular Level
We target neurological pathologies using novel genome editing techniques
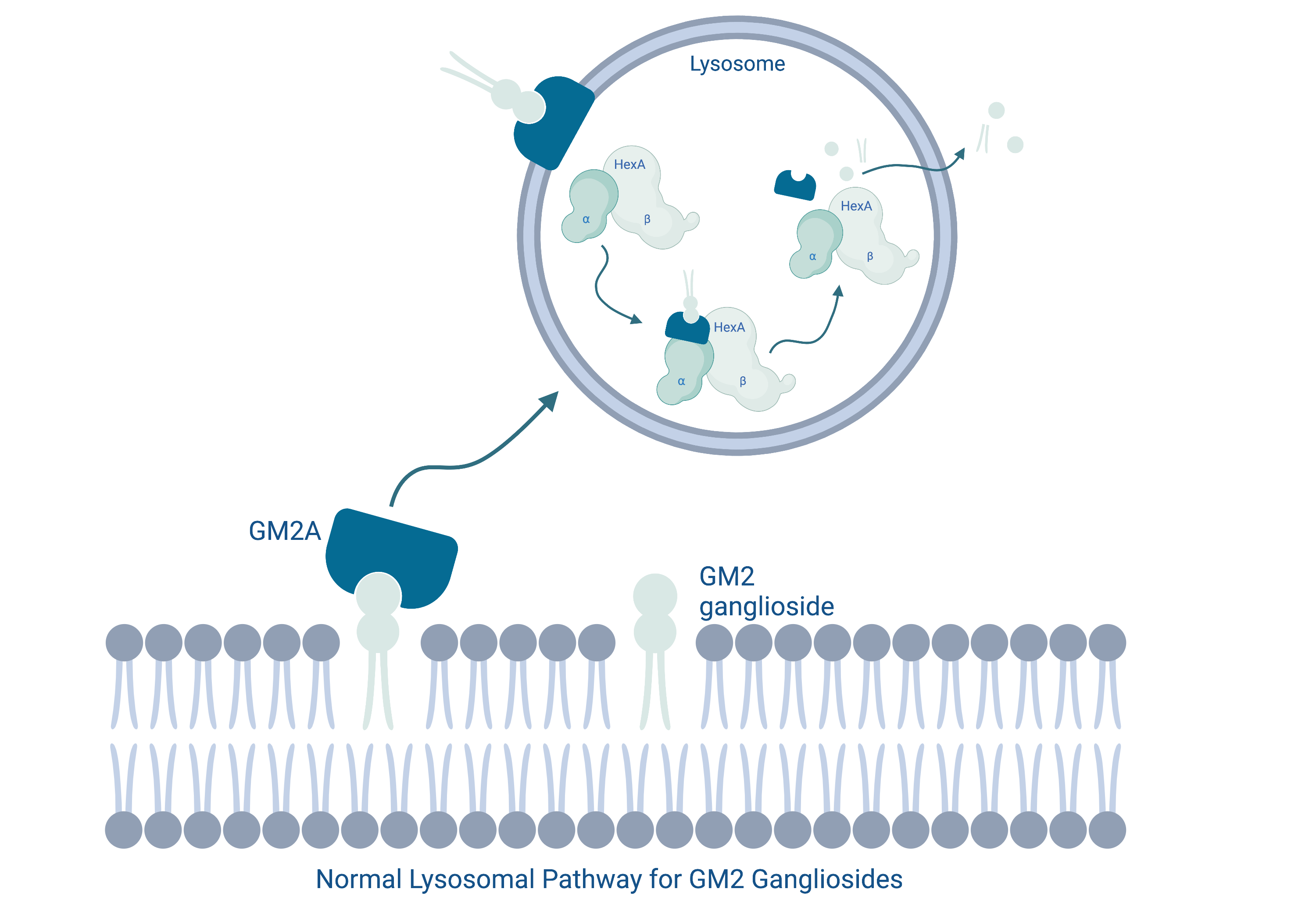
Tay-Sachs disease
Tay-Sachs disease is caused by mutations in the HEXA gene which encodes the alpha-subunit of the enzyme beta-hexosaminidase A. This is a lysosomal enzyme responsible for breaking down a cell membrane lipid called GM2 ganglioside. As a result of this mutation, the enzyme becomes dysfunctional leading to the accumulation of GM2 ganglioside, principally in central nervous system neurons. This causes neuronal dysfunction and death, which ultimately manifests as symptoms ranging from involuntary muscle twitches, vision and hearing loss, intellectual disability and seizures.
Our lab has generated a murine model of Tay-Sachs disease and is beginning to implement CRISPR-Cas 9 based therapeutics to target and correct the mutation.
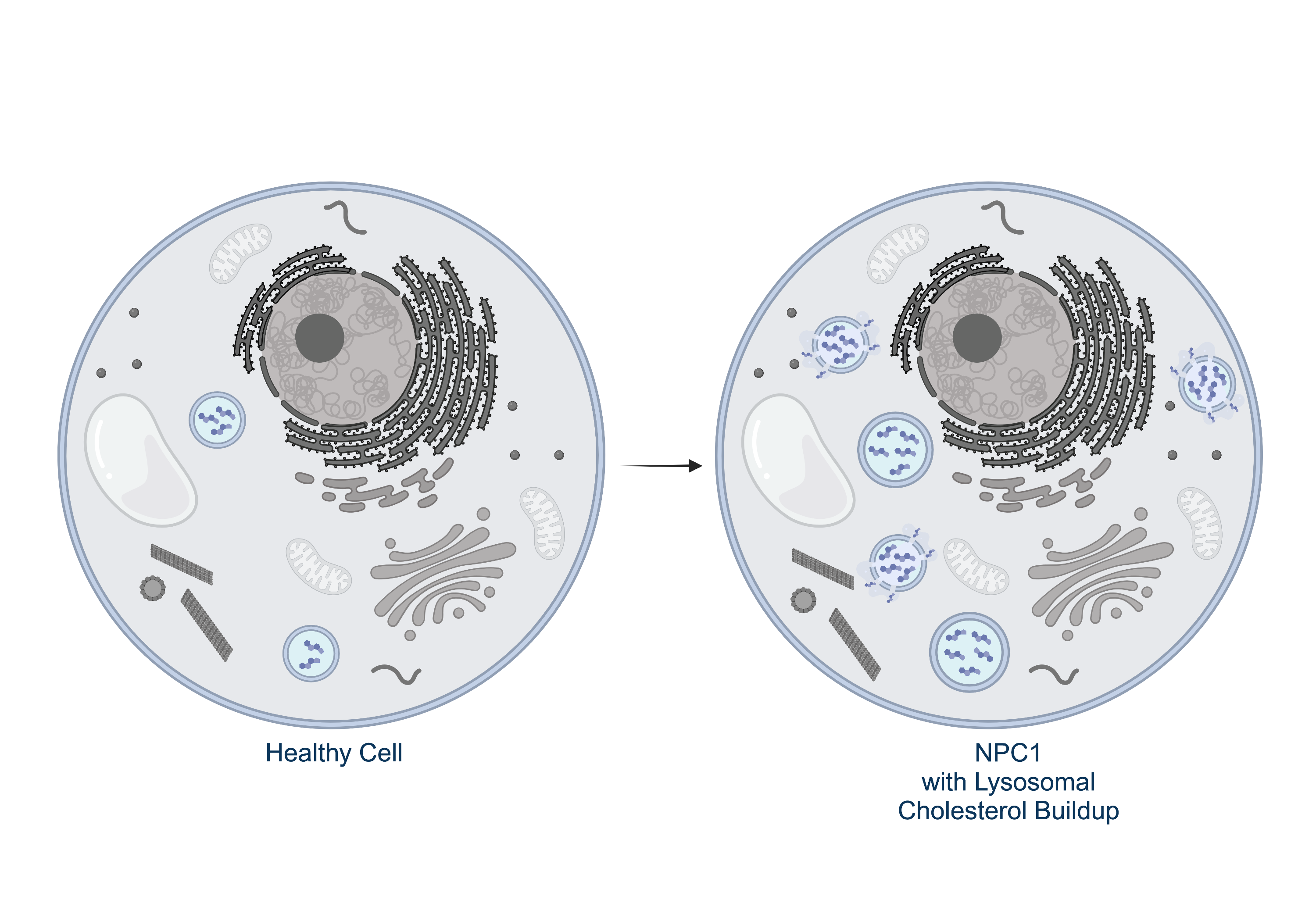
Niemann-Pick disease type C
Niemann-Pick disease type C is a metabolic disorder characterized by the abnormal accumulation of lipids, such as cholesterol and glycosphingolipids, within cells. This accumulation primarily affects the liver, spleen, and brain and is largely caused by irregularities in lysosomal storage and trafficking. The combined effects of these physiological abnormalities manifest as progressive and fatal neurodegeneration in NPC patients.
Our lab focuses on mutations in the NPC1 and NPC2 gene which play a role in lipid trafficking, transport and storage.
Progranulin-deficient frontotemporal dementia
Frontotemporal dementia is a common cause of dementia that occurs due to the damage and eventual death of neurons located in the frontal and temporal lobes of the brain. This progressive cerebral atrophy can cause changes to behaviour and personality as well as nonfluent aphasia. Our lab focuses on progranulin-deficient frontotemporal dementia, which arises due to the lack of a highly conserved protein called progranulin. Progranulin plays a role in cell growth, survival and repair and is implicated in this inherited form of dementia.
Currently, our lab is using a CRIPSR-Cas 9 to upregulate progranulin in murine models, in hopes to understand more about the disease phenotype and potential therapeutics.
Joubert-like syndrome
Joubert-like syndrome is predominantly an autosomal recessive disorder. It can be caused by mutations in several genes, and is part of a larger group of disorders called ‘ciliopathies’. Ciliopathies affect cellular structures called cilia, which are microtubule-based, cytoplasmic extensions which are critical for developmental and physiological functions. Joubert syndrome affects the brain and is characterized by the appearance of a typical ‘molar tooth sign’ on MRI. Therefore, affected individuals often display intellectual disability. Many other parts of the body can be affected as well, and symptoms can include weak muscle tone, abnormal breathing patterns, and abnormal eye movements.
Our goal is to investigate the significance of clinical variants in new genes and develop better therapeutic strategies for affected individuals.
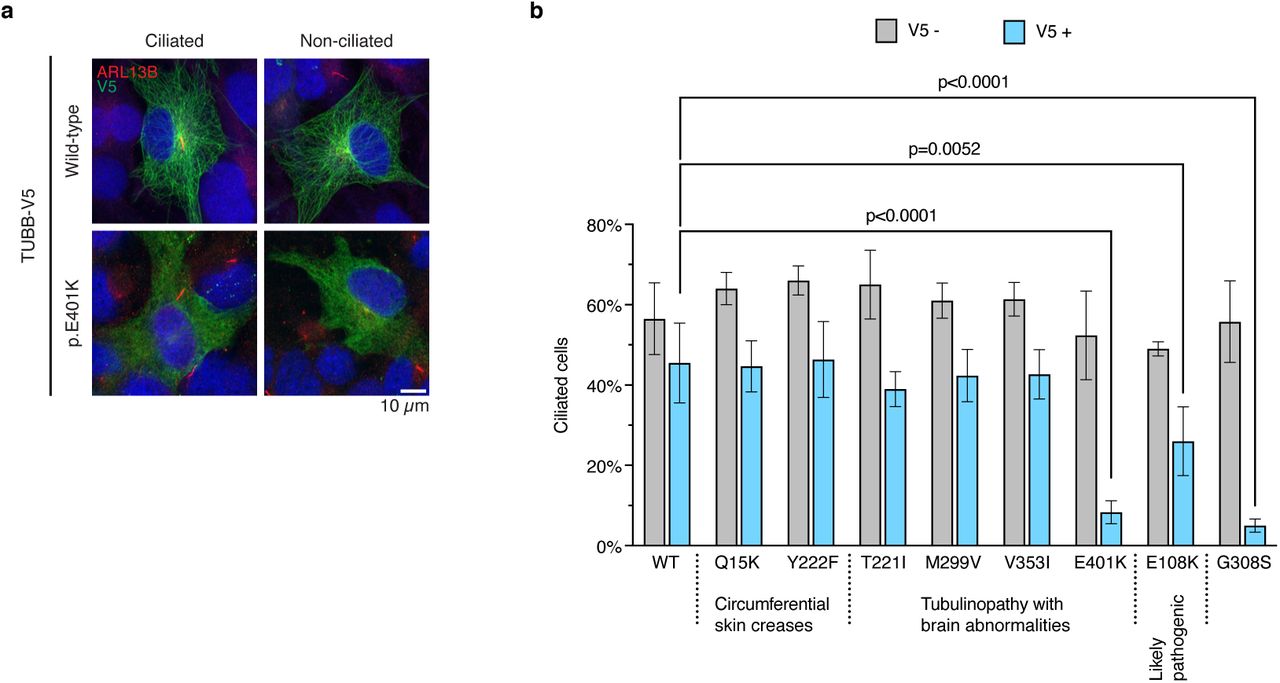
A subset of clinically relevant dominant mutations in TUBB affects ciliation in hTERT-RPE1 cells. View preprint.
MECP2 duplication syndrome
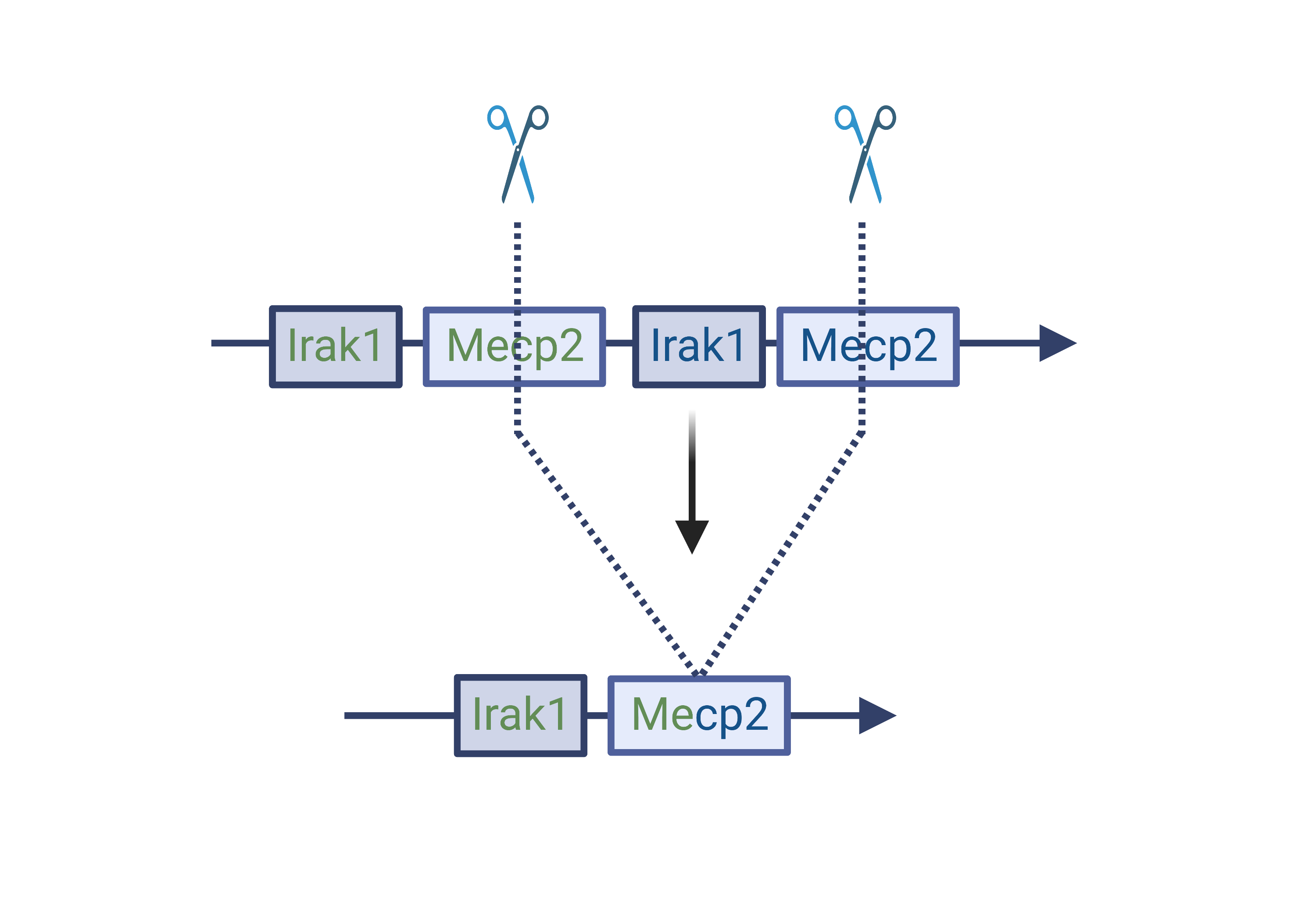
Methyl CpG binding protein 2 (MECP2) duplication syndrome (M2DS) is a rare, X-linked recessive, neurodevelopmental disorder primarily affecting young boys. The disease is caused by a duplication of the global transcriptional regulator, MECP2, and its neighboring genes. These duplications result in a pathological disruption of neuronal homeostasis by altering regulation of neuronal genes. This ultimately manifests in developmental delay, intellectual disability, autism spectrum disorder, and early death.
Our lab aims to generate, characterize and correct patient-relevant M2DS mutations in human neuronal cells to reverse the neuronal phenotypes conferred by the disease.
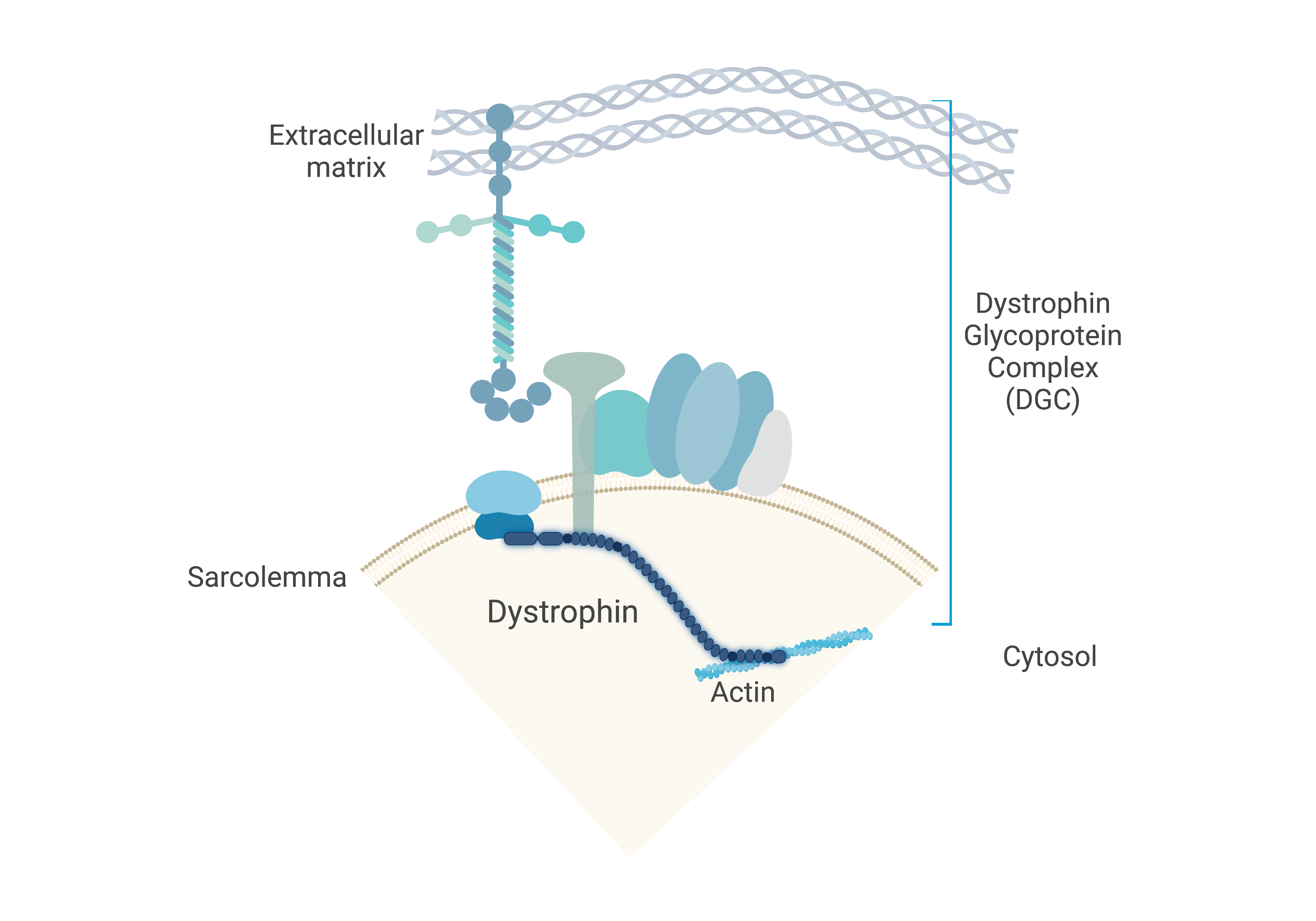
Duchenne muscular dystrophy
In collaboration with the Cohn Lab
DMD is an X-linked recessive neuromuscular disorder that affects 1 in 5,000 boys. This disease is caused by a lack of expression of the dystrophin protein, which is integral to the dystrophin glycoprotein complex (DGC) in skeletal muscle. Dystrophin creates a buffering system during cycles of muscle contraction and relaxation. Therefore, the loss of dystrophin leads to progressive muscle wasting, loss of ambulation, and cardiorespiratory complications that lead to the limited life span of these DMD patients.
Our goal is to restore either truncated or full-length dystrophin using gene editing strategies to ameliorate DMD disease phenotype.